Research
Mitigating the energy challenge is one of the most important tasks at hand for the scientific community today. One of the key ingredients to success is the design of molecularly tailored nanostructured materials that can enable alternative energy conversion and storage technologies such as solar cells and batteries. At Computational Nanoscience Laboratory, we model transport processes relevant to synthesis, processing, and performance of materials for various energy applications. The ultimate objective is to develop models that can identify materials for specific applications such as organic photovoltaic solar cells and Li-air batteries.
We use atomistic and molecular modeling techniques to investigate materials relevant to energy conversion and storage devices. Examples of current and past research efforts are provided below.
Modeling All-Solid State Sodium Batteries
All solid state sodium ion batteries, which operate at near room temperatures, have demonstrated excellent energy densities and eliminate the shortcomings of liquid electrolytes. However, developing solid state electrolyte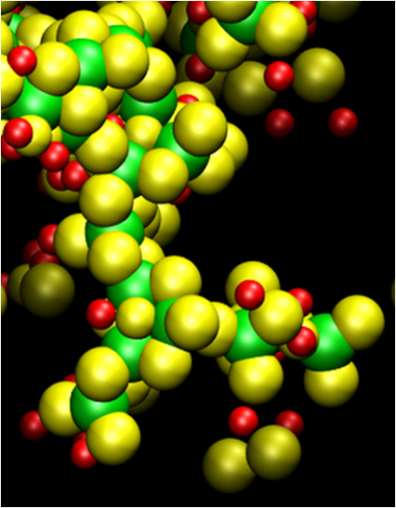 s with high ionic conductivity is a significant challenge. The current work explores sulfide glasses as potential electrolytes for all solid-state sodium batteries. Motivated by the lack of systematic theoretical evaluation of the mechanism of ion conduction, classical molecular dynamics approach was applied to evaluate the structures and ionic conductivity of a wide range of sodium sulfide – silicon sulfide [xNa2S – (1-x) SiS2] glassy electrolytes. Due to the amorphous nature of these electrolytes, various starting configurations obtained using a typical melt quench technique were explored to gather statistically reasonable structures. In order to validate the model, the results from the pair distribution functions for [0.5Na2S – 0.5SiS2] were compared with structure factor data obtained from experiments. Finally, the ionic conductivity was calculated for varying composition to identify the most promising electrolytes.
s with high ionic conductivity is a significant challenge. The current work explores sulfide glasses as potential electrolytes for all solid-state sodium batteries. Motivated by the lack of systematic theoretical evaluation of the mechanism of ion conduction, classical molecular dynamics approach was applied to evaluate the structures and ionic conductivity of a wide range of sodium sulfide – silicon sulfide [xNa2S – (1-x) SiS2] glassy electrolytes. Due to the amorphous nature of these electrolytes, various starting configurations obtained using a typical melt quench technique were explored to gather statistically reasonable structures. In order to validate the model, the results from the pair distribution functions for [0.5Na2S – 0.5SiS2] were compared with structure factor data obtained from experiments. Finally, the ionic conductivity was calculated for varying composition to identify the most promising electrolytes.
This project is funded by ![]()
Morphology of Thin Films in Perovskite Solar Cells
Over the past few years the power conversion efficiency (PCE) of planar perovskite solar cells (PSCs) has reached up to ~ 20%. However, structural and chemicals defects that lead to hysteresis in the perovskite based thin film pose challenges. Recent work has shown that thin films of PCBMs deposited on the photo absorption layer, using solution processing techniques, minimize surface pin holes 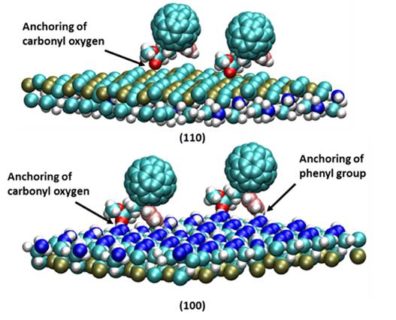 and defects thereby increasing the PCE. We have developed and employed a multiscale model
and defects thereby increasing the PCE. We have developed and employed a multiscale model 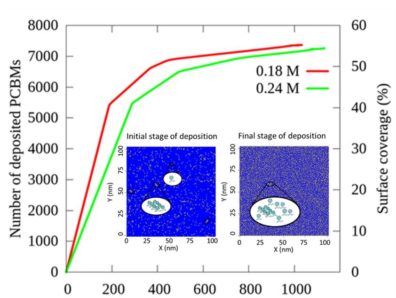 based on molecular dynamics (MD) and kinetic Monte Carlo (kMC) to establish a relationship between deposition rate and surface coverage on perovskite surface at varied PCBM concentrations. The MD simulations of PCBMs dispersed in chlorobenzene, sandwiched between (110) perovskite substrates, indicate that PCBMs are deposited through anchoring of the oxygen atom of carbonyl group to the exposed lead atom of (110) perovskite surface. Based on rates of distinct deposition events calculated from MD, kMC simulations were run to determine surface coverage at much larger time and length scales than accessible by MD alone. The outcome of the study provides a generic relationship between deposition rate of PCBMs and surface coverage of perovskite crystal and also provides detailed insights into the morphology of the deposited film.
based on molecular dynamics (MD) and kinetic Monte Carlo (kMC) to establish a relationship between deposition rate and surface coverage on perovskite surface at varied PCBM concentrations. The MD simulations of PCBMs dispersed in chlorobenzene, sandwiched between (110) perovskite substrates, indicate that PCBMs are deposited through anchoring of the oxygen atom of carbonyl group to the exposed lead atom of (110) perovskite surface. Based on rates of distinct deposition events calculated from MD, kMC simulations were run to determine surface coverage at much larger time and length scales than accessible by MD alone. The outcome of the study provides a generic relationship between deposition rate of PCBMs and surface coverage of perovskite crystal and also provides detailed insights into the morphology of the deposited film.
This project is funded by 
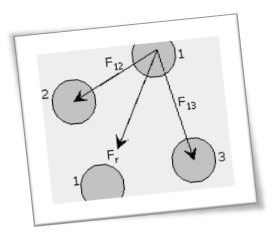
Multiscale Model to Investigate Particle Self-Assembly
Due to their wide scale applications, the aggregation mechanism and size and shape of certain colloidal particles, such as gold, silver and silica, have been widely investigated using classical and  stochastic models. These models are unable to account for atomistic scale effects and interactions of colloidal particles during clustering. We present a novel multi-scale model, based on molecular dynamics (MD) and kinetic Monte Carlo (kMC), which can both account for atomistic interactions and at the same time access the growth of colloidal particles to much greater sizes relevant to real-life applications. A well-studied system, comprising fullerenes in NaCl electrolyte solution, was simulated to validate the model. We predicted the growth mechanism of fullerene particles by analyzing the size and fractal dimensions of the growing agglomerates. Under the simulated conditions, the agglomeration process evolves from a diffusion limited cluster aggregation (DLCA) regime to percolating cluster in transition and finally to a gelation regime and shows good agreement with existing theory of colloidal particle growth. Our model shows great promise and can be utilized in myriad applications, where agglomeration of colloidal particles is of interest including solvent based processing of nanoparticles for thin film nanocomposites.
stochastic models. These models are unable to account for atomistic scale effects and interactions of colloidal particles during clustering. We present a novel multi-scale model, based on molecular dynamics (MD) and kinetic Monte Carlo (kMC), which can both account for atomistic interactions and at the same time access the growth of colloidal particles to much greater sizes relevant to real-life applications. A well-studied system, comprising fullerenes in NaCl electrolyte solution, was simulated to validate the model. We predicted the growth mechanism of fullerene particles by analyzing the size and fractal dimensions of the growing agglomerates. Under the simulated conditions, the agglomeration process evolves from a diffusion limited cluster aggregation (DLCA) regime to percolating cluster in transition and finally to a gelation regime and shows good agreement with existing theory of colloidal particle growth. Our model shows great promise and can be utilized in myriad applications, where agglomeration of colloidal particles is of interest including solvent based processing of nanoparticles for thin film nanocomposites.
Modeling-Based Design of Novel Electrolytes
Lithium ion batteries are widely used as a power source for portable devices in the consumer electronics market. However, the highest energy storage capacity achieved by a state-of-the-art Li-ion battery is too low to meet current demands in larger applications
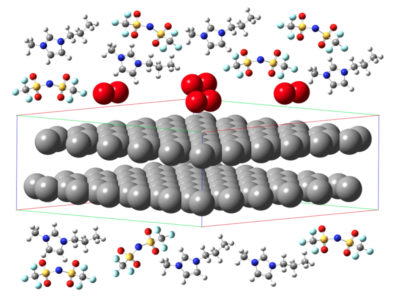 such as in the automotive industry. The limitation is due, in part, to the limited ionic conductivity of currently used organic electrolytes coupled with their volatility and flammability, which raises safety concerns. The development of new generation of Li ion batteries with significantly improved energy storage would require the selection of novel electrolyte materials with improved performance without compromising on safety standards. Room temperature ionic liquids (IL) possess unique properties, such as low vapor pressure and non-flammability, making them promising alternatives for use as Li battery electrolytes. However, IL’s often exhibit a great degree of ion association resulting in multiple anions coordinating with a single Li ion. Such enhanced coordination produces negatively charged clusters which can greatly reduce the mobility of Li and thus degrade the performance of IL electrolytes. Addition of organic diluents has been shown to enhance the transport properties of Li within IL’s. As part of this ongoing project, we are simulating IL-based electrolytes for Li batteries.
such as in the automotive industry. The limitation is due, in part, to the limited ionic conductivity of currently used organic electrolytes coupled with their volatility and flammability, which raises safety concerns. The development of new generation of Li ion batteries with significantly improved energy storage would require the selection of novel electrolyte materials with improved performance without compromising on safety standards. Room temperature ionic liquids (IL) possess unique properties, such as low vapor pressure and non-flammability, making them promising alternatives for use as Li battery electrolytes. However, IL’s often exhibit a great degree of ion association resulting in multiple anions coordinating with a single Li ion. Such enhanced coordination produces negatively charged clusters which can greatly reduce the mobility of Li and thus degrade the performance of IL electrolytes. Addition of organic diluents has been shown to enhance the transport properties of Li within IL’s. As part of this ongoing project, we are simulating IL-based electrolytes for Li batteries.
This project is funded by 
Modeling the Photoactive Layer in Organic Photovoltaic Cells
Organic photovoltaic (OPV) solar cells, which comprise photoactive layers with organic nanoparticle acceptors dispersed in conjugated polymers, have generated great scientific interest. The ease of manufacturing, relative low costs, light weight, flexibility and low operating temperature of OPVs also make them attractive candidates for commercial use. 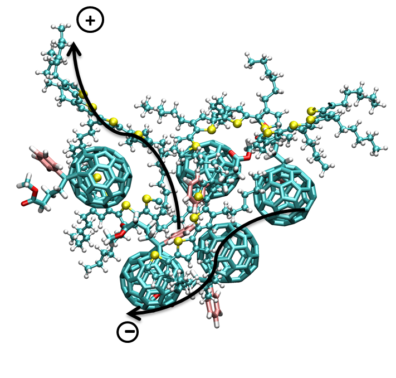 The power conversion efficiency (PCE) is still limited to less than 8-9% in OPVs due, in part, to the recombination losses of excitons. Such losses can be diminished by maximizing the contact surface area between the electron donor and acceptor materials in the photoactive layer. The photoactive layer is most often synthesized through spin coating of a solvent containing nanoparticles dispersed in a polymer. The molecular interactions between the solvent, nanoparticles and polymers during the spin coating process influence the morphology of the photoactive layer and hence determine the PCE. Prediction of optimal synthesis parameters, such as choice of solvent, processing temperature, and concentration of nanoparticles and polymers, requires fundamental understanding of the mechanisms that govern the agglomeration of nanoparticles and polymers in solvents. As part of an ongoing project, we are conducting atomistic simulations of systems relevant to OPVs. Analysis of the results from the numerical simulations will aid the selection of novel polymers and nanoparticles for OPVs.
The power conversion efficiency (PCE) is still limited to less than 8-9% in OPVs due, in part, to the recombination losses of excitons. Such losses can be diminished by maximizing the contact surface area between the electron donor and acceptor materials in the photoactive layer. The photoactive layer is most often synthesized through spin coating of a solvent containing nanoparticles dispersed in a polymer. The molecular interactions between the solvent, nanoparticles and polymers during the spin coating process influence the morphology of the photoactive layer and hence determine the PCE. Prediction of optimal synthesis parameters, such as choice of solvent, processing temperature, and concentration of nanoparticles and polymers, requires fundamental understanding of the mechanisms that govern the agglomeration of nanoparticles and polymers in solvents. As part of an ongoing project, we are conducting atomistic simulations of systems relevant to OPVs. Analysis of the results from the numerical simulations will aid the selection of novel polymers and nanoparticles for OPVs.

This project is funded by
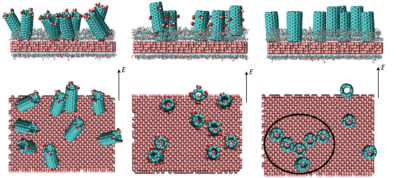
Controlled Deposition of Aligned Carbon Nanotubes on Substrates
Self-assembly of carbon nanotubes (CNTs) on substrates has myriad applications including nanotube based electrochemical energy conversion and storage devices, such as batteries and super-capacitors. Several such applications require deposition of vertically 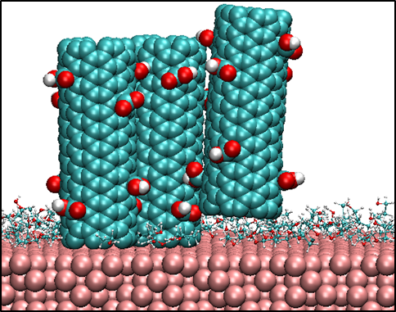 or horizontally aligned CNTs on substrates in order to control the effective electrical conductivity and mechanical properties of these devices and achieve substantial improvement in their performance. Solution-processed deposition of CNTs provides a cost-effective means to synthesize uniform vertically or horizontally aligned nanostructures on top of substrates. The efficacy of deposition depends on the solubility of CNTs in the solvent as well as the ordering of nanotubes relative to the substrates. These governing factors, which determine the specific morphologies of CNTs that are deposited, are governed by the molecular interactions between the CNTs and the substrate and solvent molecules. In an effort to mimic the conditions during solution-processed deposition of nanotubes on substrates, we are conducting atomistic simulations to study systems comprising carbon nanotubes and commonly used solvents sandwiched between substrates. Analysis of the results from the simulations will provide fundamental insight to predict key factors for solution-processed deposition of well-ordered CNTs on substrates.
or horizontally aligned CNTs on substrates in order to control the effective electrical conductivity and mechanical properties of these devices and achieve substantial improvement in their performance. Solution-processed deposition of CNTs provides a cost-effective means to synthesize uniform vertically or horizontally aligned nanostructures on top of substrates. The efficacy of deposition depends on the solubility of CNTs in the solvent as well as the ordering of nanotubes relative to the substrates. These governing factors, which determine the specific morphologies of CNTs that are deposited, are governed by the molecular interactions between the CNTs and the substrate and solvent molecules. In an effort to mimic the conditions during solution-processed deposition of nanotubes on substrates, we are conducting atomistic simulations to study systems comprising carbon nanotubes and commonly used solvents sandwiched between substrates. Analysis of the results from the simulations will provide fundamental insight to predict key factors for solution-processed deposition of well-ordered CNTs on substrates.