Exotic Clusters
Understanding how the step-wise addition of atoms leads to the transition from a single atom to a diatomic molecule to an atomic cluster and finally to the formation of bulk solid allotropes represents a fundamental problem. Experimental and theoretical analyses can elucidate the structural and electronic changes that occur during the assembly of individual atoms and how they form atomic clusters and bulk materials of interest. Even something as small as the addition or removal of a single atom (either guest or host atom) in the endohedral clusters may have a profound effect on their overall geometrical structures (see [Nb@Ge13/14]3– and [Pt1/2@Sn17]4– clusters shown in the figure below), leading us to explore the causes of such drastic alterations. The geometric changes could also be accompanied by significant electronic structure changes; impacting their guest-host chemical bonding and affecting their stability and reactivity. In our research, we aim to identify the correlations between the geometric and electronic structures that ultimately will help us rationally design materials with tailored physical and chemical properties. In these projects, we closely collaborate with the inorganic chemist Prof. Zhong-Ming Sun from Nankai University who synthesize these exotic clusters as cryptand salts.
![Cage structure of niobium-germanium clusters. [NbGe13]3-. [NbGe14]3-. Cage structure of Platinum Tin clusters. [Pt2@Sn17]4-. [Pt@Sn17]4-](https://s3.wp.wsu.edu/uploads/sites/3458/2024/09/geometry-changes-396x463.png)
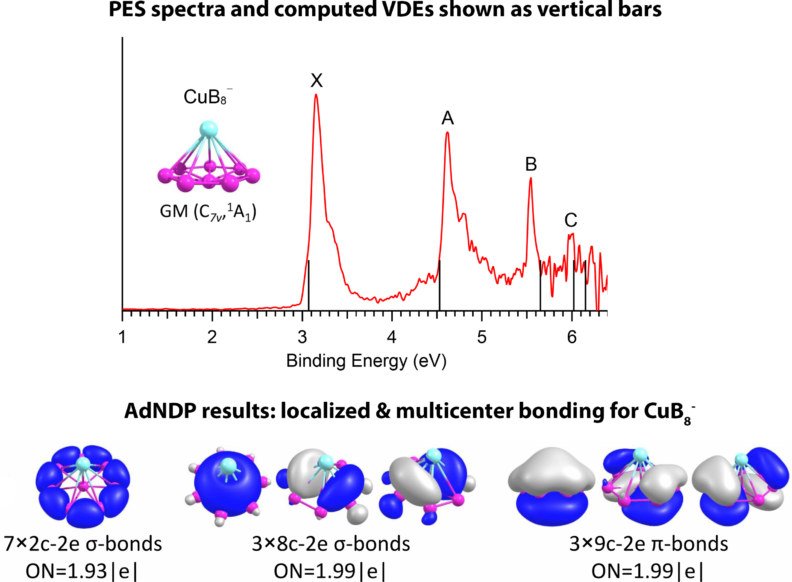
While XRD data analysis is typically employed to determine the geometry of the clusters made in a solid state, there is no structural information for the gas-phase clusters produced in anionic photoelectron spectroscopy (PES) experiments. To determine the geometry of the cluster, it is required to employ a global minimum (GM) energy search to identify the most stable structure on the potential energy surface of a specific stoichiometry, charge, and spin state. For this purpose, we employ the Coalescence Kick (CK) algorithm, which includes several steps: (1) random positioning of atoms within a designated Cartesian box, (2) gradually moving these atoms towards the center of mass until a pre-defined threshold parameter is met, (3) performing a local optimization with a chosen DFT method, and (4) iteratively executing steps 1 through 3, hundreds of thousands of times. The application of the CK algorithm is beneficial because it allows us to identify both the GM structures and their corresponding low-lying isomers. In our lab, we collaborate with photoelectron spectroscopist Prof. Lai-Sheng Wang from Brown University, who records PES for various pure and doped boron clusters to systematically investigate their structures and properties. We simulate the PES peaks by calculating vertical detachment energies (VDEs) corresponding to the removal of an electron from a specific orbital, e.g., HOMO (VDE1), HOMO–1 (VDE2), HOMO–2 (VDE3), etc. (see figure below showing the PES for CuB8– cluster). To understand why the cluster of interest adopts a specific geometry and how atoms are bonded, we perform chemical bonding analyses by employing various electron localization methods. The lower panel of the figure below shows the results from the Adaptive Natural Density Partitioning analysis for the GM structure of the CuB8– cluster.