Theoretical modeling of bonding in lanthanide and actinide systems is very challenging. In strongly correlated systems with a partially filled f-shell, Coulombic repulsion tends to localize individual f-electrons at the atomic sites while hybridization with the ligand orbitals tends to delocalize them. Actinide-ligand and lanthanide-ligand interactions are the most complex and polytypic in nature due to the increased nodality arising from the availability of the multiple lobes in the f-orbitals. Canonical molecular orbitals are typically very delocalized and difficult to interpret in terms of traditional Lewis bonding. Plus, the metal atomic contributions may be overly spread (especially in the high oxidation states of the metal) that makes it difficult even to identify the number of unpaired electrons on the metal.
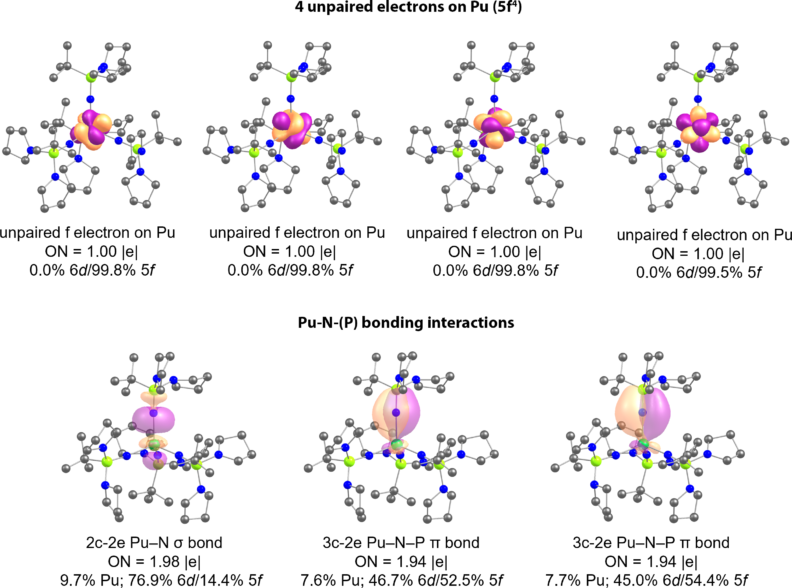
In our research, we employ electron localization algorithms, orbital and energy decomposition analyses, and molecular orbital energy level diagrams to disentangle the complex electronic structure of f-block compounds. In particular, we are interested in the mechanisms of covalency in actinide systems, investigating both orbital overlap and orbital energy degeneracy. Adaptive Natural Density Partitioning (AdNDP) method may provide a total orbital contribution of the actinide metal in the An–L interactions as well as information on the polarization of the bonds and their hybridization (e.g. 6d vs. 5f An contributions). At the same time, AdNDP delineates the metal contributions to the localized non-bonding elements from those describing An–L bonding, providing information about the lone pairs or unpaired electrons on An (figure below shows AdNDP results for the Pu4+ imidophosphorane complex synthesized by our collaborator Prof. La Pierre from Georgia Institute of Technology).
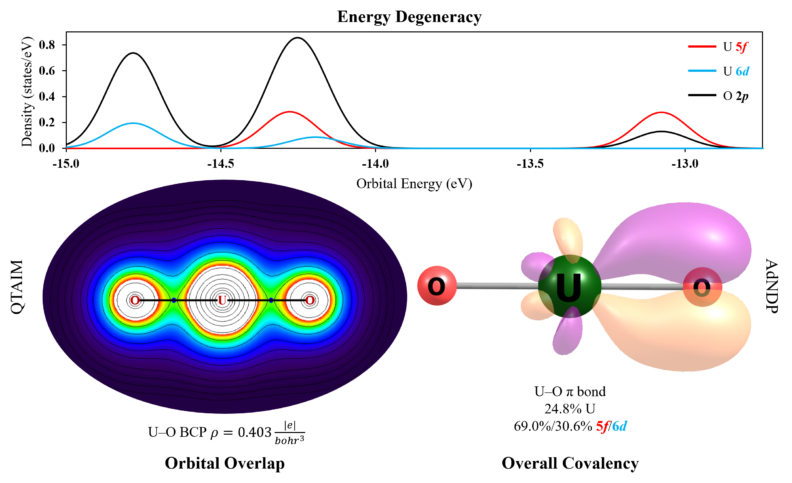
Another convenient way to assess the degree of covalency of a bond is through topological analysis of the electron density (ρ) via the quantum theory of atoms in molecules (QTAIM). QTAIM provides quantifiable value of ρ at the bond critical point (BCP) given by the Hamiltonian matrix element (HML), which can be used to determine the significance of orbital overlap. In addition, the delocalization index (DI) can serve as a parameter to assess the collective effect of the orbital overlap and energy degeneracy of the metal and ligand orbitals (E0M and E0L). In particular, the orbital energy degeneracy can be tracked via the density of states plots, as exemplified below for the [UVIO2]2+ molecule, displaying the energy match between actinide 5f and 6d orbitals with valence O 2p orbitals and the density of those orbitals within a given energetic range. To elucidate these quandaries, the Popov lab uses several methods which function together to paint a coherent picture of chemical bonding and the role of 5f/6d orbitals in actinide systems to guide experimental design of ligands that capitalize on such properties.
Overall, assessment of the degree of localization/delocalization, polarization, hybridization, and covalency of the An–L interactions is crucial for the rational design of An compounds. We are particularly interested in understanding how the electronic structure of transuranic elements changes going from one extreme to another, from low to high oxidation states. In our research, we aim to identify the changes of covalency that could be tracked in the overall reactivity, redox properties, and thermodynamic stability. This is an important piece of information that can be leveraged to develop better separation methods and for the design of quantum information technologies. On one hand, we are thrilled to elucidate highly reduced species, like AmII compounds, which should be stable because of their 5f7 electronic structure, but still have not been experimentally observed. Understanding this anomaly from the theoretical perspective will lead to a better understanding of the chemistry of Am and other actinides in general. On the other hand, we aim to develop theoretical guidelines that could be used to design non-actinyl transuranic coordination compounds in high oxidation states, like AmVI, wherein the metal orbitals are expected to mix with the ligand orbitals significantly, thus imposing a challenge for the description of their electronic structures.
