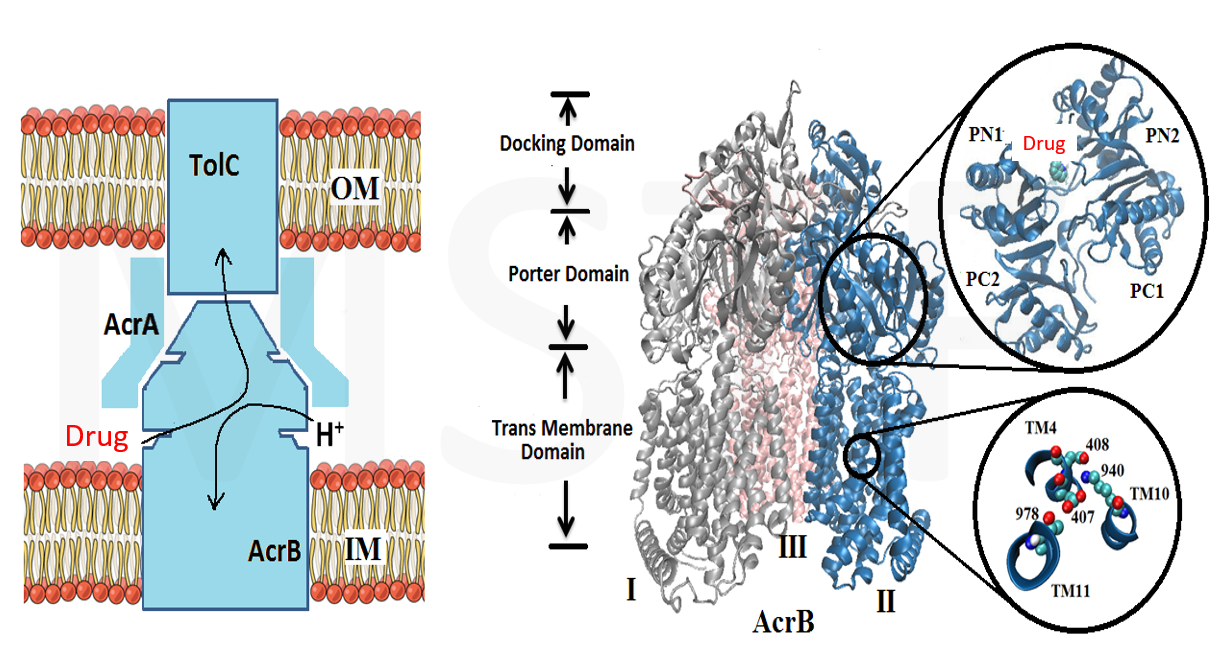
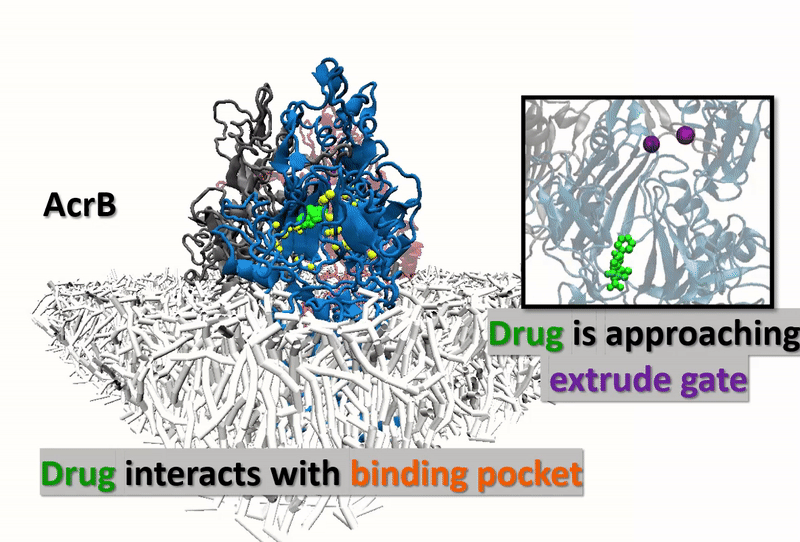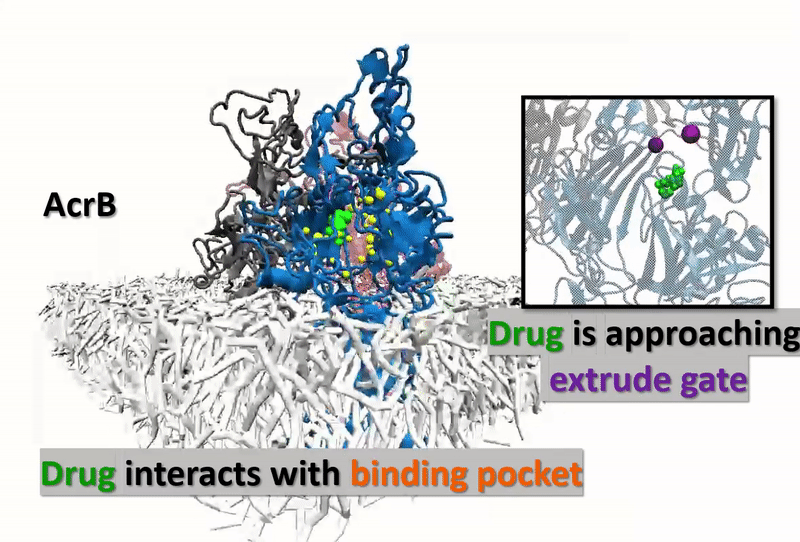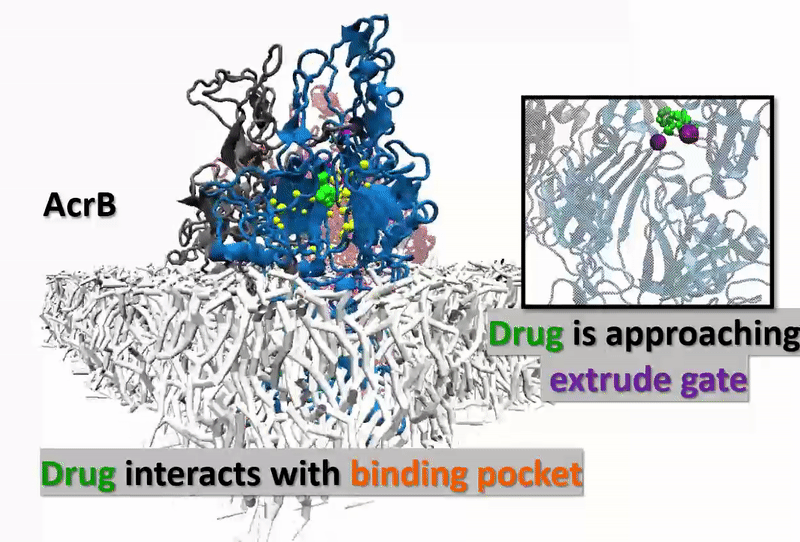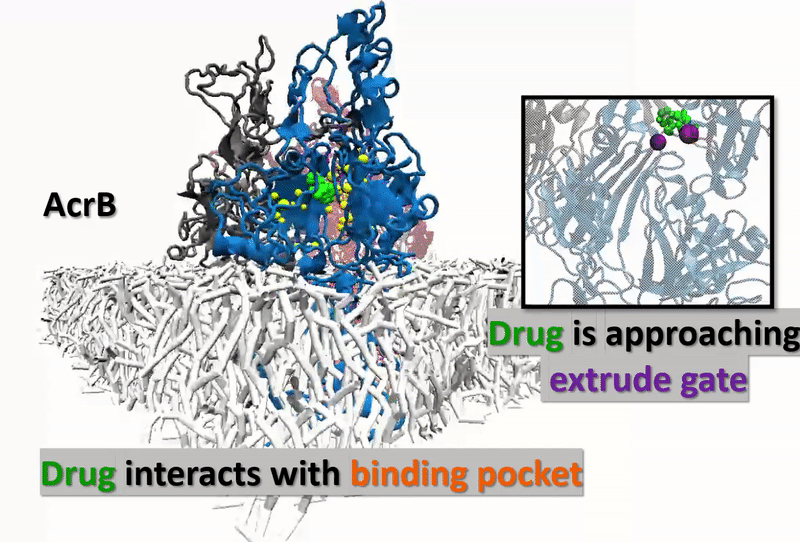
The multidrug resistance (MDR) system defensively excreted anti-infection agents out of cells causing extreme medical issues. AcrB (one of the key segments of MDR) experiences a progression of vast scale and proton-rationale conformational changes. Catching the conformational changes through all-atom simulation is difficult and time-overwhelming. Here, we actualize a united-atom coarse-grained force field to explore the conformational changes of AcrB in the porter domain under various protonation conditions of Asp407/Asp408 in the trans-membrane area. Drug behavior testing is then investigated. We, at the first step, developed united-atom PACE force field for variation of selective drug those are referred to have strong and special interactions with AcrB. The numerical experiments next aim to investigate how the drugs react with AcrB binding pockets as well as how AcrB monomers respond back to the drug appearances. The results could be the first time, show drugs’ impact on protonation conformations of AcrB, obstructing the drug-excretion process.
Our developed force-field data gives accurate agreement with other experimental and computational references. Thus, the approach opens a big opportunity in observing a micro-seconds simulated process (which is arduously captured by using experiments or all-atom MD simulations) in molecular-biotic systems. AcrB, with the drugs inside, shows the different response of the drug entrance/exit gates compared with the without-drug AcrB system. The final goal of this research is supposed to open wider computational manner in the drug testing industry, not only with AcrB but also many other significant biotic-systems.